Kliknij ikony powyżej, aby pobrać najnowsze ApE (v2.0.61, Februrary 5, 2020)
Zobacz poniższe instrukcje dotyczące instalacji programów open source na komputerach Mac. Jeśli instalujesz na OSX El Capitain (OSX 10.11) lub starszych systemach. Wyślij mi wiadomość e-mail z prośbą o instrukcje (proszę podać wersję systemu operacyjnego – wiadomości bez tej informacji będą ignorowane.)
Kliknij tutaj , aby przekazać dobrowolną darowiznę na rzecz ApE.
Śledź @ApEplasmid
Krótki film o instalacji ApE na komputerze Mac:
Twoja przeglądarka nie obsługuje znacznika <video>.
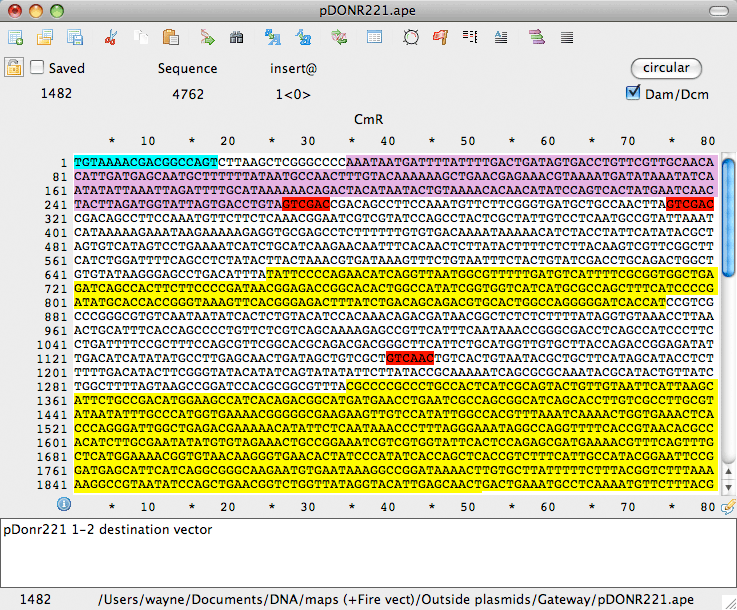
Działa w systemach Windows (XP, Vista, 7, 8, i 10) i Mac (OS X v10.5 i wyżej) Podświetla miejsca restrykcyjne w oknie edycji Dokładnie odzwierciedla blokowanie Dam/Dcm miejsc enzymatycznych Podświetla tekst przy użyciu predefiniowanych i własnych bibliotek cech Pokazuje translację, Tm, %GC, ORF wybranego DNA w czasie rzeczywistym Odczytuje pliki DNA Strider, Fasta, Genbank i EMBL Zapisuje pliki jako kompatybilne z DNA Strider lub w formacie Genbank Podświetla i rysuje mapy graficzne używając adnotacji cech z plików genbank i embl Bezpośrednio BLASTuje wybraną sekwencję w NCBI lub wormbase
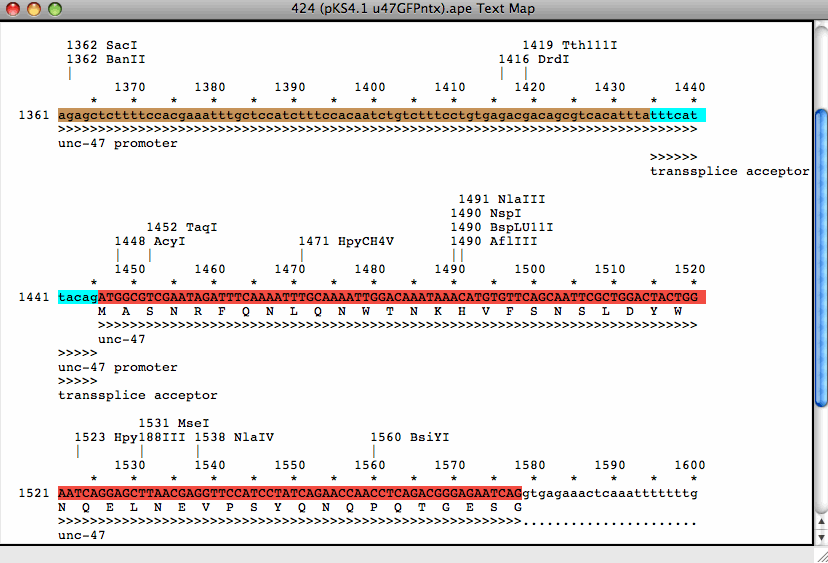
Mapa tekstowa pokazuje sekwencję DNA, translację, i cechy jako grafika tekstowa
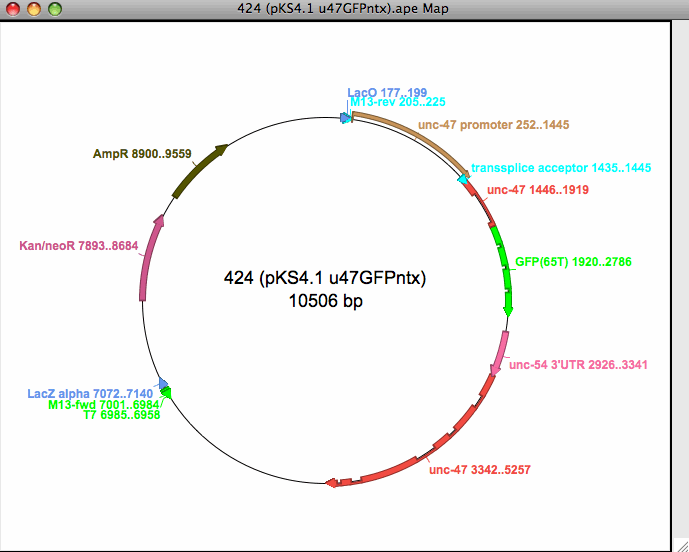
Tworzy graficzne mapy restrykcji- liniowe lub kołowe z zaznaczonymi cechami. Liniowe lub kołowe z zaznaczonymi cechami Łączy cechy graficzne i tekstowe za pomocą podwójnego kliknięcia hiperłącza Zapisuje grafikę jako enkapsulowany postscript lub skalowalną grafikę wektorową Kopiuje i zapisuje grafikę jako metapliki Windows (tylko MS Windows)
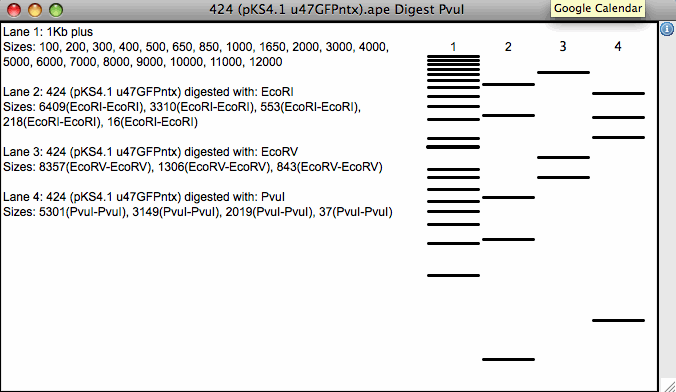
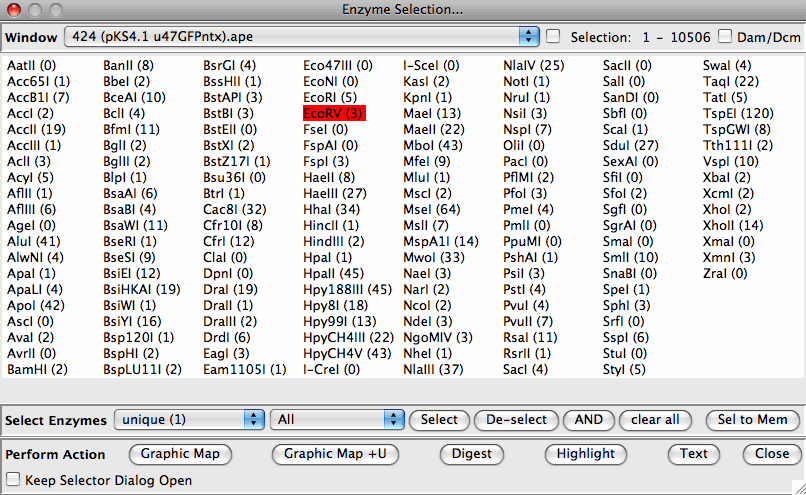
Wirtualne trawienie restrykcyjne Rysuje wstępnie
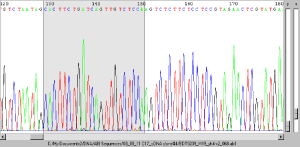
Odczytuje pliki śladów sekwencjonowania ABI Sekwencje w śladach ABI można wyrównać bezpośrednio do sekwencji referencyjnej, z wyrównaniem połączonym hiperłączem z powrotem do śladu.
Wybiera miejsca pasujące do wielu kryteriów (unia/przekrój – częstotliwość cięcia, typ miejsca) we wszystkich otwartych oknach Wybiera miejsca, które są cięte częściej w jednej sekwencji niż w innej (do wykrywania snip-SNP lub digestów diagnostycznych) Ma zdefiniowane przez użytkownika grupowanie enzymów w celu wyróżnienia np. enzymów aktualnie dostępnych w magazynie. Umożliwia użytkownikom definiowanie nowych enzymów według nazwy i miejsca rozpoznania Importuje pliki w formacie DNA Strider (proste enzymy, listy miejsc) dostępne w REBASE
Inne funkcje:
Większość okien analizy jest połączona hiperłączami z odpowiadającymi im sekwencjami, w tym: Mapy graficzne Mapy tekstowe Wirtualne digesty Alignacje (w tym sekwencje ABI) Silent Sites Translation Primer Find Wykorzystuje niestandardowe biblioteki definicji cech, które pozwalają na: Szybką adnotację sekwencji Szybkie wyszukiwanie i podświetlanie wszystkich dostępnych primerów, które posiadasz (lub innych), które hybrydyzują do sekwencji Sekwencja może być adnotowana i wizualizowana na wiele sposobów szybko i wydajnie Mapy graficzne, które pokazują miejsca wiązania primerów i wszystkie interesujące cechy sekwencji Tłumaczy sekwencje z opcjonalnym wyrównaniem DNA Znajduje potencjalne primery pasujące do kryteriów użytkownika (długość, Tm, %GC, komplementarność własna/inna) Dopasowuje dwie sekwencje DNA (lub dowolną kombinację sekwencji i śladu ABI), z dopasowaniem połączonym hiperłączem do oryginalnej sekwencji Znajduje nieme translacyjnie miejsca restrykcyjne Rysuje graficzne mapy ORF
Kliknij tutaj aby przekazać dobrowolną darowiznę na wsparcie ApE.
Pobierz starsze wersje
Możesz teraz bezpośrednio eksportować regiony genomowe z Wormbase: W prawym górnym przycisku menu rozwijanego wybierz „Download Track Data”. Kliknij przycisk „Konfiguruj…”. Zrzuć wybrane cechy używając wersji 3 Across aktualnie widocznego regionu. Zaznacz „Zapisz do pliku”. Zaznacz „Osadź sekwencję DNA”. Odznacz „Uwzględnij dane konfiguracyjne śladu”. Kliknij „Go”. Otwórz wynikowy plik .gff w najnowszym ApE.
Alternatywnie, możesz wyeksportować region genomowy (z przeglądarki genomu) jako plik w formacie FASTA (używając menu w lewym górnym rogu). Można dodać ścieżki cech, pobierając pliki ścieżek cech GFF3 za pomocą tego samego menu. W ApE otwórz plik FASTA, a następnie użyj menu Features, aby otworzyć informacje o ścieżkach GFF3.
Innym sposobem jest pobranie modelu genu (ze strony genów), wklejenie go do okna ApE, a następnie zaznaczenie wszystkich, utworzenie nowej cechy (menu Feature), a w oknie edycji cechy, które się pojawi naciśnij przycisk „tylko duże litery”. Jeśli uważasz, że ApE nie znajduje wszystkich miejsc ClaI (lub XbaI lub BclI), które WIESZ, że są obecne w twojej sekwencji, wyłącz metylację Dam/Dcm na twojej sekwencji i spróbuj ponownie. Zobacz tutaj po więcej informacji. Kliknij tutaj , aby przekazać dobrowolną darowiznę na rzecz ApE.


 , aby przekazać dobrowolną darowiznę na rzecz ApE.
, aby przekazać dobrowolną darowiznę na rzecz ApE.